







Theoretical Projects
A mean-field approach for the determination of the polarizabilities
for the water molecule in liquid state
(with P. G. KUSALIK, Department of Chemistry, University of Calgary, AB Canada)
for the water molecule in liquid state
(with P. G. KUSALIK, Department of Chemistry, University of Calgary, AB Canada)
A mean-field method is presented describing the electrostatic environment experienced by water molecule in liquid state, which is used to extract the corresponding hyper- and high-order polarizabilities. Within this approach, MD computer simulations of liquid water samples for two standard water potentials at several different temperatures are performed to characterize the distributions (specifically average values) of local fields and field gradients. The electric response properties (including non-linear contributions up to fourth-order) are then calculated using ab initio techniques in conjunction with a charge perturbation variant of a finite field method. Sets of fixed charges are used to generate the desired electric fields and electric field gradients. Calculations of dipole polarizability, hyper- and principal components of high-order polarizabilities of the water molecule in gas and liquid phase conditions are carried out at MP2 and MP4levels of theory; the values obtained for three different liquid phase models are compared with those for gas phase. For a liquid phase water molecule the first hyperpolarizability (β) and first higher polarizability (A) increase markedly, actually changing sign. The second hyperpolarizability (γ) also increases but much less dramatically, and components of the second high-order polarizability tensor (B) demonstrate a rearrangement of contributions. We observe that a less symmetrical gradient model gives the most accurate representation of liquid-phase conditions. The excellent agreement of our gas-phase values with experimental results and the most accurate previous theoretical predictions is evident of the quality of our higher order polarizabilities and theoretical models.

The total molecular dipole moment for liquid water
(with P. G. KUSALIK, Department of Chemistry, University of Calgary, AB Canada)
(with P. G. KUSALIK, Department of Chemistry, University of Calgary, AB Canada)
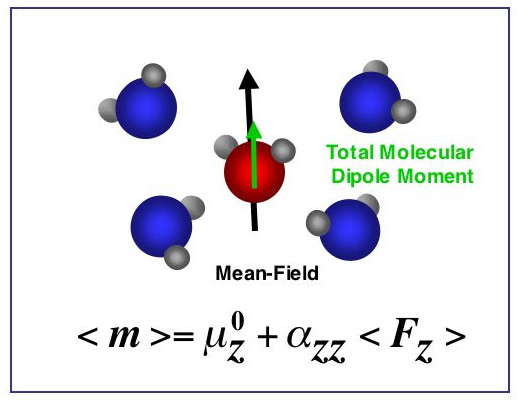
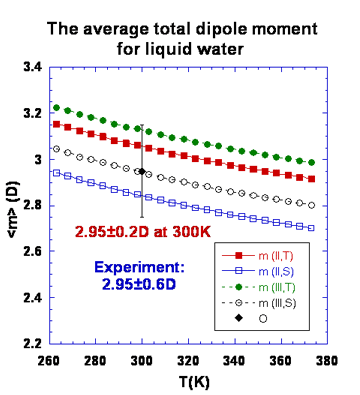
For the water molecule, the dipole is the first nonzero multipole moment; it represents the polarity of the molecule and has been widely used in describing solvation behavior. A rather wide range of theoretically determined values for the total molecular dipole moment of water in condensed phases has been reported in the literature. This paper describes a means by which the average total dipole moment for the water molecule in the liquid state can be linked to experimental refractive index data. Three components comprise the mean-field approach that is employed. A formal framework is developed that relates the temperature dependence of the effective molecular polarizability to the average local electric field experienced by a liquid water molecule over a chosen temperature range. A characterization of the distributions of local fields and field gradients is also necessary, and this has been determined from the computer simulations of liquid water samples at several different temperatures for two standard water potentials. The final component, the electric response properties of the water molecule (including nonlinear contributions up to fourth order), were determined from ab initio calculations for gas- and liquid-phase molecules, and are reported in A. V. Gubskaya and P. G. Kusalik, Mol. Phys. 2001, 99, 1107. By combining these three components, the temperature dependence of the average local electric field, and consequently the average total dipole moment, are extracted from data for the refractive index of liquid water. An almost 10% variation in the dipole moment with temperature is observed over the range 273 to 373 K. The value obtained for the molecular dipole moment at 300 K, 2.95±0.2 D, is in excellent agreement with a recently reported result extracted from x-ray scattering data, as well as with some recent theoretical predictions.
 |
 |
Molecular dynamics simulations of 1,2-disubstituted (hydroxy- and amino-) ethanes:
Pure molecular liquids and their aqueous solutions
(with P. G. KUSALIK, Department of Chemistry, University of Calgary, AB Canada)
Pure molecular liquids and their aqueous solutions
(with P. G. KUSALIK, Department of Chemistry, University of Calgary, AB Canada)
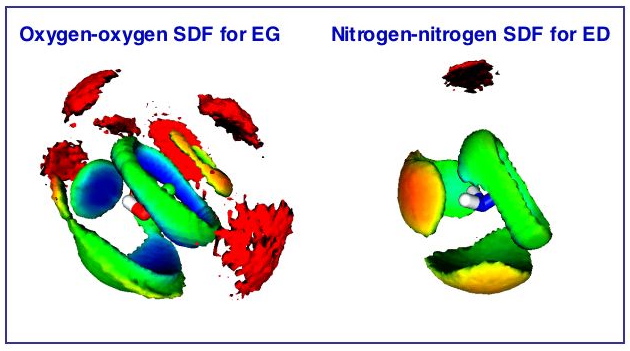
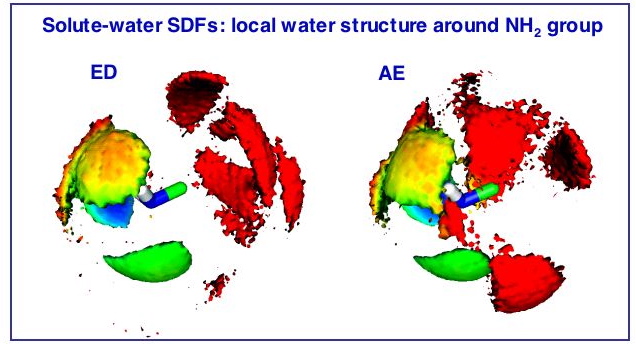
This project is a comparative computational study of the local structure of three widely used representatives of 1,2-disubstituted ethanes, namely ethylene glycol (EG), ethylenediamine (ED) and 2-aminoethanol (AE), in liquid state and their mixtures with water. Classical molecular dynamics combined with three-dimensional atomic density maps, known as spatial distribution functions (SDF’s), are the computational tools used in this study. The present work consists of three parts. In the first part, twelve molecular models were designed and gas-phase simulations were carried out for each. The results obtained were compared with the most reliable experimental estimates in order to test different force fields and molecular representations. In the second part liquid-phase simulations were performed on the most successful (AMBER/OPLS-based) models. The heats of vaporization and self-diffusion coefficients were used as criteria for the final selection of molecular models to be employed in the subsequent simulations of aqueous solutions. In the third part, a detailed structural analysis was performed. As an essential part of this analysis the dihedral angle distributions were calculated and relative populations of conformers with respect to the central dihedral angle were determined for pure EG, ED and AE, as well as their mixtures with water, where four compositions of each compound were considered. It has been confirmed that in the liquid phase the gauche conformation accounts for the major population of rotational isomers for EG and AE, while ED exhibits a significant population of trans conformers. Additionally, the first theoretical estimates of the compositional dependence of self-diffusion coefficients for the aqueous solutions of EG, ED and AE were obtained. The analysis of radial distribution functions in conjunction with calculated numbers of nearest neighbors around oxygen and nitrogen atoms of the main functional groups provided some structural insights into the H-bonding pattern of the systems studied. The number of strongly H-bonded neighboring groups was determined and their possible positions were located by means of SDF’s. The possibility of four-membered H-bond arrangements (comprised of two strong and two weak H-bonds) found around oxygen and nitrogen atoms leads to the conclusion that in the liquid phase the generalized H-bonding pattern for EG, ED and AE can be described as a three-dimensional, branched network.
 |
 |